
How unique are feral honey bee populations: Using genetics to characterize feral honey bee colonies
How unique are feral honey bee populations: Using genetics to characterize feral honey bee colonies
Project Overview:
The United States has no native honey bees. Its resident honey bee populations originate from centuries of importation from Europe, Africa, and Asia. At least eight subspecies have been imported into the country over the last 400 years. Honey bees in their native range have varying tolerance to Varroa and its associated viruses, they express different behaviors, and they are favored differently by beekeepers. This is also true in their introduced range in the United States but here, honey bees are a genetic mix of subspecies brought over from the native range. Our labs have recently discovered that feral honey bees—those which live outside of typical managed apiaries—may be more tolerant to viral diseases than managed colonies. This observation suggests that there are genetic differences between feral and commercial populations that result in differences in viral tolerance. However, it is entirely unclear if a genetically distinct group of feral populations of bees exists within the US. We have already created a large database of genomic sequence for commercially-available honey bee stocks within the United States. We proposed to expand this data set to feral colonies and quantify genetic differentiation between feral populations and commercially available stocks. We were able to identify uniquely adapted and genetically distinct populations of feral honey bees within the US which can be protected and incorporated into breeding schemes to help beekeepers maintain healthier stocks.
Milestones:
We collected foraging honey bees from across the states of Indiana, Kentucky, and Pennsylvania, and North Carolina (Midwest and Northeast of the US). Our sampling was impeded by the COVID19 pandemic, but we successfully sourced feral and commercial colonies from the above locations. We obtained genomic sequencing data from colonies in feral and commercial colonies across each state and used a Pool-Seq approach to quantify genetic diversity, differentiation, and ancestry across both the genome (Figure 1)
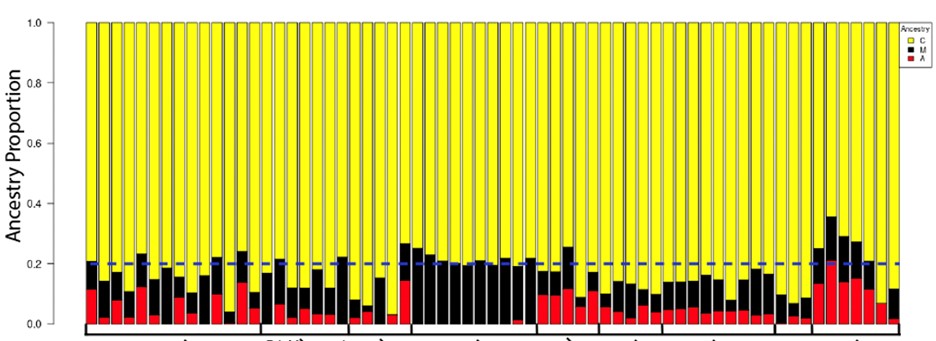
Figure 1. Ancestry plot of selected honey bee genomes collected across the North America by PD Harpur (selected from 150 total samples). Each color represents the fraction of genomic ancestry originating from each of three ancestral source populations. Ancestry is largely of C-lineage origin but there is significant variation in ancestry across populations. This variation is directly associated with phenotypic variation across the country. (red: A lineage; yellow: C lineage; black: M lineage).
We found significant differences in the ancestry and genetic differentiation of some feral populations relative to commercial populations. Our results suggest this difference is driven by the level of isolation a feral population experiences. We are currently preparing this section of the results for publication. We were able to generate three peer-reviewed papers with these generous funds. We were able to completely document the movements of honey bees into the United States (Carpenter & Harpur, 2021) and we were able to improve the honey bee genome (Kaskinova et al. 2021).
We feel that these results provide an important resource for beekeepers and scientists by identifying reservoirs for local adaptation and genetic diversity. We know feral honey bee populations have been lost to disease and habitat loss in the US. By identifying reservoirs of genetic diversity, we can better protect them in the future. All of this is vital to the long-term success of honey bees in North America. We feel our proposal meets a pressing need for the beekeeping community and it continues Eva Crane’s remarkable work in understanding the consequences of human management on honey bees.
Related Papers:
Kaskinova M, Yunusbayev B, Altinbaev R, Raffiudin R, Carpenter MH, Kwon HW, et al. Improved Apis mellifera reference genome based on the alternative long-read-based assemblies. G3. 2021;11. doi:10.1093/g3journal/jkab223
Carpenter MH, Harpur BA. Genetic past, present, and future of the honey bee (Apis mellifera) in the United States of America. Apidologie. 2021. doi:10.1007/s13592-020-00836-4
Expected Outcomes:
What allows feral colonies to survive when infected with viruses? Several explanations exist. First, feral colonies may have different levels of genetic diversity than commercial colonies. Higher genetic diversity at the colony level has been associated with tolerance to viral disease (Desai & Currie 2015). Second, feral populations may have experienced natural selection to be resistant to viruses. If viral tolerance is heritable and feral colonies are more likely to mate with other feral colonies, then selection may have favored viral tolerance in feral populations. Finally, there may be environmental differences (diet or colony conditions) that contribute to viral tolerance in feral colonies (Dolezal et al. 2019). Testing these three hypotheses would contribute greatly to our understanding of how bees become virus tolerant and to our ability to help our bees fight off viral infections. For example, support for the first two hypotheses implies that bee breeders could incorporate ‘feral genetics’ into their breeding schemes and select for viral tolerance. Support for the third hypothesis implies that management practices can be changed to increase honey bee viral tolerance.
Here we propose to quantify genetic differences among feral and commercial honey bee populations across the United States. We propose to collect honey bee samples from State and National forests within the US. We will directly test the two genetic hypotheses proposed above: that feral honey bees have different levels of genetic diversity than commercial stocks and they are genetically distinct from commercial stocks. Our labs have already compiled the largest available genomic data set for commercial honey bee populations (Carpenter et al; in prep) allowing us to directly compare genetic diversity and divergence between feral populations to commercial.
Specifically, we propose to:
- Collect feral honey bees from populations across Midwestern and Northeastern United States.
- Sequence the genomes of collected samples and quantify genetic diversity within each sampling location
- Compare feral genomic data to known commercial data sets to directly determine differences in genetic diversity and estimate divergence.
Brock Harper
Dept. Entomology
Purdue University