
The genomic basis of adaptive sub-species divergence in Apis mellifera.
The genomic basis of adaptive sub-species divergence in Apis mellifera.
Background
Beekeepers have long asserted that honey bee sub-species possess different traits that are indicative of adaptation to their local environmental conditions. While there are many excellent studies published relating to genetic lineages and honey bee diversity, systematic studies into these sub-species traits, and whether they have an underlying genetic basis, are lacking. This is because gathering these data demands both replicated, large-scale experimental studies of co-located colonies of each sub-species, and genomic-scale colony data to identify any phenotypic differences and their genotypic associations. To-date there has been no such large-scale field studies due to the logistic challenges of carrying that out, and similarly no parallel genomic study due to the lack of subjects and the technical challenges that, until relatively recently, were beyond the scope of most studies.
Understanding the genetic signatures underlying phenotypic characteristics in different A. mellifera sub-species is important for several reasons. Along with the significant advantage of informing current breeding practices and enhancing our understanding of evolutionary processes, it also enables a comprehensive database of genome diversity in these sub-species to be catalogued, understood and actively conserved, at a time when it is very rapidly being lost. The practice of honey bee transhumance across the European continent has resulted in introgression (the transfer of genetic variation [alleles] between species by repeated backcrossing) of honey bee sub-species. This has the potential to disrupt genetic combinations that have been acquired over long time periods as a result of adaptive evolution. In Germany, for example, the human mediated movement of honey bees has resulted in native Apis mellifera mellifera being entirely replaced due to commercial interests and the perceived desirable characteristics of non-native sub-species.
Our study
This study is the result of a collaboration between the University of Plymouth as part of Victoria Buswell’s PhD project, for which Prof. Mairi Knight is the primary supervisor, and a research project based in Sweden and Norway (“Supporting Nordic Brown Bees, a Unique Resource for our Ecosystem Services” led by Drs Mats Niklasson, Tora Vinterli Hoel and Sonja Leidenberger and funded by the EU via INTERREG Swede-Norway European Regional Development Fund).
The Nordic INTERREG project’s aims were to investigate sub-species trait differences between the major European sub-species of honeybee (four in total: Apis mellifera mellifera, A. m. carnica, A. m. ligustica and the hybrid ‘Buckfast bee’) through exactly such a replicated, large- scale experiment, using co-located colonies of each subspecies at a number of different sites, as described above. The Eva Crane Trust then funded the University of Plymouth team to collaborate with the INTERREG project researchers to carry out the genomic screening of each colony.
The experimental work involved monitoring 5 apiaries and a total of 78 colonies (see Fig 1). Data were gathered by the Nordic INTERREG research team on a number of colony characteristics including parasite load, pollen and honey characteristics, behaviour (e.g. defence, guarding, swarming, etc.), colony weight, life history (e.g. winter survival, flight activity) and environmental variables (light intensity, temperature, rainfall, wind speed and direction).

Figure 1. a) Dr Sonja Leidenberger (right) and Victoria Buswell (left); b) Sonja indicating some of the data logging equipment installed in each colony, both photographs were taken at one of the experimental apiaries near Uddevalla in southern Sweden.
For the genomic screening, previous work (Buswell, submitted) had established the importance of, in this context, of screening multiple individuals from each colony to ensure the collective genotype is captured (since queens mate multiply). Thus, 30 workers from each colony were sampled and pooled so that the full genotypes of each colony could be established. Seventy-one colonies belonging to the three different sub-species and one hybrid line (Buckfast) were genotyped and compared to a set of sub species standards (n= 7 A.m.mellifera; n = 7 A.m. carnica; n= 5 A.m.m.ligustica, (with thanks to Gabriele Soland, Apigenix, Switzerland; Gabel Martin, Landesbetrieb Landwirschaft Hessen, Germany and Raffaele Dall’Olio, Agricultural Research Council, Italy). Samples were prepared in the laboratory at the University of Plymouth followed by whole-genome-sequencing (BGI, Hong Kong).
Key findings
After stringent filtering for quality control, 208,587 single nucleotide polymorphisms (SNPs) were extracted from the pooled colony genomes for downstream analysis. The first task was to establish the genomic identity of each colony compared to the standards, and to cross-match that to the putative sub-species assignment in the experiment. Note that the Buckfast colonies were excluded from these analyses, leaving n=51 colonies. ADMIXTURE and PCA analysis confirmed that overall species assignment was good, with several colonies within each putative sub-species emerging as ‘pure’ (with no signatures belonging to the other two sub-species). However, it also identified varying levels of introgression in some of the experimental colonies of all three sub-species (Figs 2, 3). These results are important since they have been fed in to the INTERREG project to inform the analysis of functional trait differences between the sub-species.
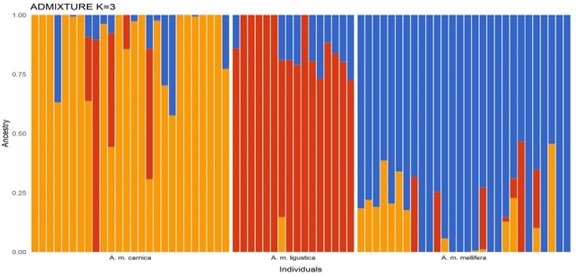
Figure 2. ADMIXTURE analysis K=3 of 208,587SNPs across 70 colonies. Here, the blue cluster represents A. m. mellifera, the red cluster represents A. m. ligustica and the yellow cluster represents A. m. carnica.
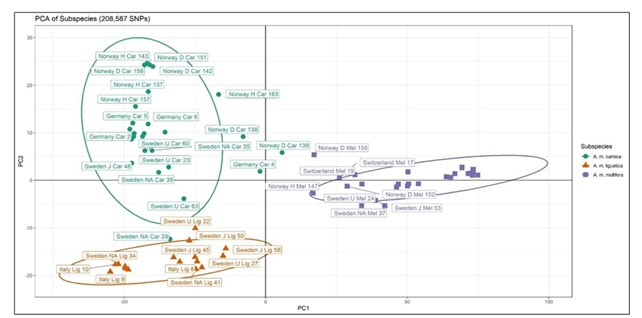
Figure 3. Principal component analysis from Adegenet. PC1 explains 43.1% of the variance while PC2 explains 2.7% of the variation in the data set. The putative subspecies is represented by colours and shapes. A. m. carnica is represented by green circles, A. m. ligustica by orange triangles and A. m. mellifera by purple squares. Samples are circled with a group ellipse with a confidence level of 0.95.
Future work
Further analysis of the SNP dataset investigated which genomic sites contributed most strongly to sub-species identity. Several such sites were identified (above the 99% percentile line in Fig. 4) distributed largely evenly across the genome, although we note a potential particularly high number on chromosome 11. These SNPs fell into different classes: those that are synonymous or neutral and are unlikely be associated with functional traits (unless through linkage), and those that indicate resent functional genomic differences. The latter category are sites that are of particular interest in terms of the second part of this planned work, which is to test which (if any) genomic sites associate with particular phenotypic traits (as collected in the experimental work). This work remains to be completed since we are waiting for the results of the INTERREG project to be collated and made available (a considerable exercise involving multiple partners and a very large and diverse dataset). While we regret that we have been unable to conduct this second important aspect of the analysis in time for this report, as we already have the genomic data this will be carried out as soon as those data become available; our collaborators in Sweden have confirmed that they expect this to be available within the coming months.
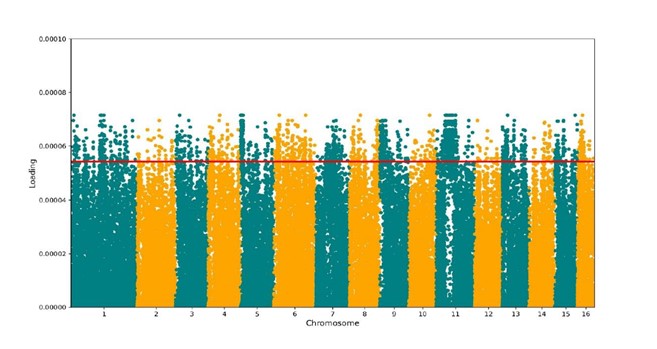
Figure 4. Genome wide visualisation of the contributions of all 208, 587 SNPs to the structure seen in the Discriminant Analysis of principal components. The x-axis represents the position of each SNP on the chromosomes and the y-axis the loading contribution for each SNP. The red line indicates the 99th percentile.
Benefit to the researchers
The Nordic INTERREG project benefitted by the genomic data usefully informing the initial A.mellifera sub-species colony assignment, and by adding the significant additional element of the genomic data to investigate the underlying genetic basis of any phenotypic differences observed. The University of Plymouth benefitted by enabling the PhD project of Victoria Buswell to achieve some of its initial goals by incorporating phenotypic data with genomic screening, something that had proved difficult to achieve through the initially proposed approach of beekeeper questionnaires. We expect the final full marriage of the results from both studies to significantly advance the field. Both parties also significantly benefitted from the opportunity to meet and work with new international collaborators interested in similar research questions.
We would therefore like to sincerely thank the Trust for funding this project, and for their patience as this work has been delayed due to the covid pandemic. While partially complete, the work remains very positively ongoing. We expect to be publishing more information as the full phenotypic dataset becomes available for further analysis.
Mairi Knight and Victoria Buswell
University of Plymouth.

